Importing sequencing data
Download
The import_seq script encapsulates all operations required to manage the high-throughput sequencing data. It will:
- Download the data from a remote location such as a sequencing center,
- Format the data to comply with a common format. This includes by default renaming files and compressing the FASTQ files with a more efficient algorithm than Gzip.
- Input the sequencing run(s) into the database.
The data (FASTQ files) are downloaded into the folder /data/seq/raw/LABXDB_EXP in the following example. By default, import_seq will download data in folder with current date.
Imported data were published in Vejnar et al. For this tutorial, only a few reads were kept per runs to reduce the file sizes.
import_seq --bulk 'LABXDB_EXP' \
--url_remote 'https://labxdb.vejnar.org/doc/databases/seq/import_seq/example/' \
--ref_prefix 'TMP_' \
--path_seq_raw '/data/seq/raw' \
--squashfs_download \
--processor '4' \
--make_download \
--make_format \
--make_staging
The sequencing runs will receive temporary IDs prefixed with TMP_. To import multiple projects, different prefixes such TMP_User1 or TMP_User2 can be used.
If you didn’t install Squashfs, you can:
- Remove
--squashfs_downloadoption and no archive.sqfs will be created and download directory will remain or, - Replace
--squashfs_downloadwith--delete_downloadoption to delete the non-FASTQ files.
After import
After completion, sequencing data can be found in /data/seq/raw:
/data/seq/raw
└── [ 172] LABXDB_EXP
├── [4.0K] archive.sqfs
├── [ 254] resa-2h-1_R1.fastq.zst
├── [ 271] resa-6h-1_R1.fastq.zst
├── [ 254] resa-6h-2a_R1.fastq.zst
└── [ 348] resa-6h-2b_R1.fastq.zst
- FASTQ files were compressed with Zstandard.
- Remaining files (all but FASTQ files) were archived with Squashfs.
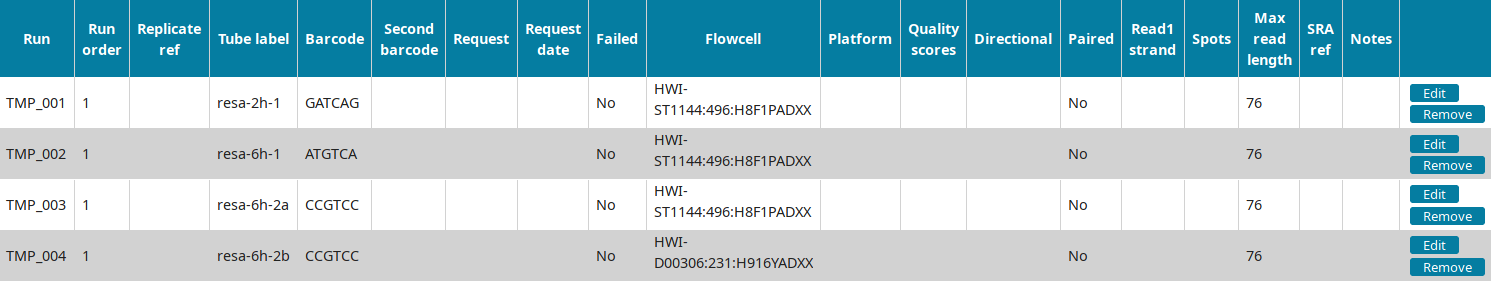
In the Run view of LabxDB seq, the 4 runs were imported:
You can then proceed to Annotating sequencing data.